
Modeling PROTAC Degradation Activity with Machine Learning
Published in Artificial Intelligence in the Life Sciences, 2024
In this work, we propose an efficient and open-source machine learning strategy to predict the degradation activity of PROteolysis TArgeting Chimeras (PROTACs).
Abstract
PROTACs are a promising therapeutic modality that harnesses the cell’s built-in degradation machinery to degrade specific proteins. Despite their potential, developing new PROTACs is challenging and requires significant domain expertise, time, and cost. Meanwhile, machine learning has transformed drug design and development. In this work, we present a strategy for curating open-source PROTAC data and an open-source deep learning tool for predicting the degradation activity of novel PROTAC molecules. The curated dataset incorporates important information such as \(pDC_{50}\), \(D_{max}\), E3 ligase type, POI amino acid sequence, and experimental cell type. Our model architecture leverages learned embeddings from pretrained machine learning models, in particular for encoding protein sequences and cell type information. We assessed the quality of the curated data and the generalization ability of our model architecture against new PROTACs and targets via three tailored studies, which we recommend other researchers to use in evaluating their degradation activity models. In each study, three models predict protein degradation in a majority vote setting, reaching a top test accuracy of 80.8% and 0.865 ROC-AUC, and a test accuracy of 62.3% and 0.604 ROC-AUC when generalizing to novel protein targets. Our results are not only comparable to state-of-the-art models for protein degradation prediction, but also part of an open-source implementation which is easily reproducible and less computationally complex than existing approaches.
Keywords: PROTAC, Machine Learning, Targeted Protein Degradation, Drug Discovery, E3 Ligase, SMILES, Deep Learning
Introduction and Motivation
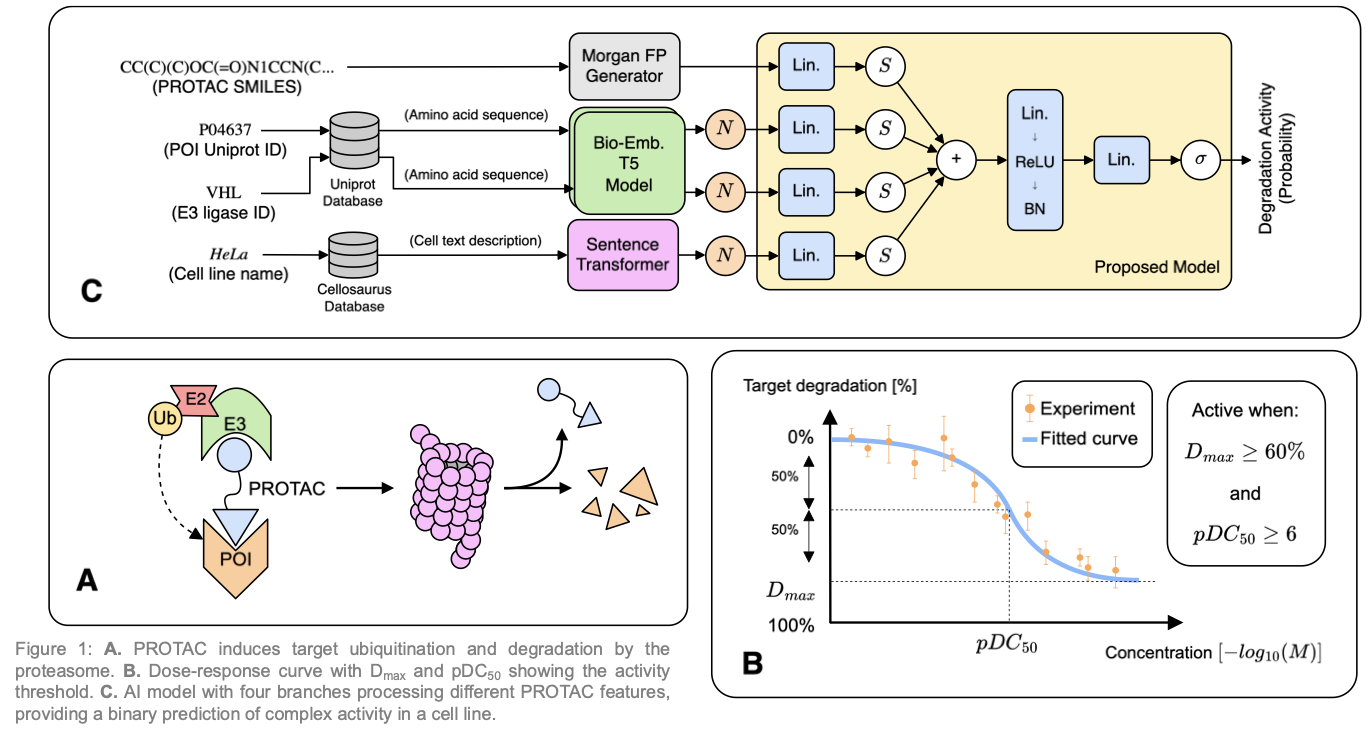
Machine learning (ML) has profoundly impacted drug design and discovery. ML efficiency can be leveraged in the development of PROTACs (PROteolysis TArgeting Chimeras), Figure 1A, a novel class of therapeutic agents that use the cell’s proteasome to degrade specific proteins, offering a promising strategy for addressing diseases with previously “undruggable” targets. However, a notable gap exists in open-source tools and resources for PROTAC research, hindering further exploration and development in this innovative therapeutic area.
Methods
Our methodology, Figures 1B-C, for predicting PROTAC-induced target degradation combines data preprocessing, feature extraction, and advanced model training, with an emphasis on specialized validation techniques:
- Data Preprocessing: Involves curating a dataset from PROTAC-DB [1] and PROTAC-Pedia [2];
- Feature Extraction: Uses protein and cell line embeddings, and computes molecular fingerprints from SMILES;
- Specialized Cross Validation: Focuses on generalization across new targets and SMILES, using a distinct test set to evaluate the model’s predictive accuracy in novel scenarios.
This approach ensures our model is well-equipped to predict with high accuracy across diverse chemical structures and biological targets.
Data Curation and Representation
We curated data from the PROTAC-DB [1] and PROTAC-Pedia [2] open-source databases, focusing on key features such as PROTAC structures (represented as SMILES), E3 ligase types, protein of interest (POI) sequences, cell line information, and degradation metrics like \(pDC_{50}\) and \(D_{max}\). We further standardized cell line names using the CelloSaurus database and encoded POI sequences with protein embeddings. The curated dataset serves as the foundation for training predictive models.
Model Architecture
The proposed model, Figure 1C, integrates multiple data representations:
- PROTAC Representation: Utilizes molecular fingerprints derived from SMILES strings to capture structural information.
- E3 Ligase and POI Encoding: Employs pretrained embeddings to represent protein sequences, capturing functional and structural characteristics.
- Cell Line Information: Encodes textual descriptions of cell lines into numerical embeddings using sentence transformers, providing context about the experimental environment.
These representations are processed through separate neural network branches, and their outputs are concatenated and passed through additional layers to predict degradation activity as a binary classification task.
Evaluation Strategy
To assess the quality of the data encoding and the model performance, we designed three evaluation studies:
- Standard Split: Randomly divides the dataset into training and test sets to evaluate an upper bound performance.
- Target Split: Ensures that POIs in the test set are not present in the training set, assessing the model’s ability to generalize to unseen targets.
- Similarity Split: Selects test set PROTACs based on their structural dissimilarity to those in the training set, evaluating the model’s capacity to predict activity for novel chemical structures.
Results
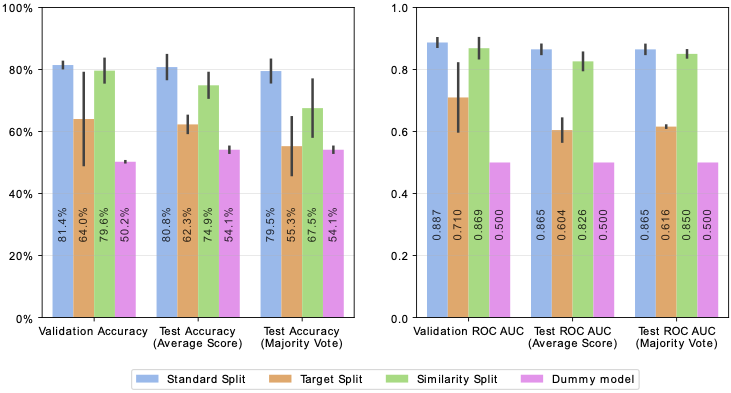
In Figure 2, our model demonstrates strong capability in predicting PROTAC-induced protein degradation (80.8% test accuracy), excelling particularly when predicting activity of molecules never encountered at training time (74.9% test accuracy). It however struggles when generalizing to new targets (62.3% test accuracy), suggesting the need for more advanced protein encodings.
Conclusion
We propose an open-source dataset curated from PROTAC- DB [1] and PROTAC-Pedia [2], setting a new foundation for future innovations in the field. Our simpler model surpasses the state-of-the-art DeepPROTACs [3], achieving a test accuracy of 80.8% versus 77.95%. Our results highlight the need for more advanced protein encodings and expanded datasets, pushing the boundaries of current methodologies.
References
[1] G. Weng, C. Shen, D. Cao, et al., “PROTAC-DB: an online database of PROTACs,” Nucleic acids research
[2] PROTACpedia - Main. [Online]. Available: https://protacpedia.weizmann. ac.il/ptcb/main (visited on 05/26/2023).
[3] F. Li, Q. Hu, X. Zhang, et al., “DeepPROTACs is a deep learning-based targeted degradation predictor for PROTACs,” Nature Communications
Code
Explore the full repository here and test out our Gradio app for interactive predictions here.
Recommended citation: Ribes, S., Nittinger, E., Tyrchan, C., Mercado, R., "Modeling PROTAC degradation activity with machine learning", Artificial Intelligence in the Life Sciences 2024